Research
Throughout my research career I have focused on combining experiments in natural and constructed populations with high-throughput genetic, genomic, and transcriptomic analyses to better understand the current and historical forces that have shaped the evolution of traits, and using those patterns to infer what changes may occur in the future. Below, I summarize some of the projects that I have led over the past ten years, in collaboration with talented undergraduate and graduate students, as well as faculty both in person and remotely at institutions in the US, Canada, Mexico, and China.
My full list of publications is available on my Google Scholar page.
Detailed analysis notebooks for all of these projects are available on my GitHub repositories.
High throughput genome sampling for trait mapping and evolutionary analysis
The cost of obtaining genetic sequence data that covers an entire genome has dramatically decreased over the past two decades. However, the work that is involved in taking the fragmented pieces of those data and putting them together into a complete, high-quality genome assembly remains complicated and expensive. Yet a completely polished genome sequence is often unnecessary to answer many interesting questions, such as what genes are responsible for different physical or behavior traits, what historical forces have shaped the current traits of organisms and how might they respond to change in the future, and what are the historical patterns of population size, migration, and composition, that may be evident in the genome?
In collaboration with researchers in four institutions in the US, and three institutions in China, I developed novel targeted sequence capture genotyping arrays that allow us to consistently obtain DNA sequences from specific regions of the genome. We have begun using these resources to generate large genomic datasets from hundreds of individuals in order to generate genetic trait maps, and to understand the evolutionary relationships among plant species that have puzzled researchers for hundreds of years.
Where are the genes that cause differences between males and females?
 Flowers of male and
female trees of the balsam poplar.
Flowers of male and
female trees of the balsam poplar.
Species that have separate male and female individuals (as opposed to species in which all individuals are hermaphrodites) often have specialized chromosomes in their genomes that guide the development of sexual traits. We often think that the sex-determining genes on these specialized chromosomes should change very rarely over time because their function is very important for successful reproduction. While some groups of organisms such as mammals and some birds do show near evolutionary stasis of sex-determining genes, other species have much more dynamically evolving sex chromosomes. Willows and poplars are plant species that are particularly interesting in this regard because they share an origin of sex chromosomes, and yet the genomic location of the mechanisms through which sex is determined appear to change frequently among these species. One of our major research goals is to identify the location and mechanisms of sex-determining genes in a large number of willow and poplar species so that we can assess the differences and commonalities in their mechanisms, as well as understanding the evolutionary forces that drive the movement of these genes.
 A sex-linked region
for the black willow on Chromosome 7. The dots represent genetic markers from
the sequence capture data on each of the 19 chromosomes, and the position of
the dot on the Y-axis represents the degree to which that dot is associated
with differences between males and females. The red-dashed line is the
significance cut-off, dots above that line are significantly different between
males and females.
A sex-linked region
for the black willow on Chromosome 7. The dots represent genetic markers from
the sequence capture data on each of the 19 chromosomes, and the position of
the dot on the Y-axis represents the degree to which that dot is associated
with differences between males and females. The red-dashed line is the
significance cut-off, dots above that line are significantly different between
males and females.
In a recent paper, available as a pre-print on bioRχiv I report the implementation of one of the targeted sequence capture arrays that I designed to identify a region on Chromosome 7 of the black willow that shows significant differences between males and females (a characteristic of sex determination genes). This project involved sequencing samples of the genome of 24 male and 24 female trees. I developed a high-throughput data analysis pipeline to analyze the resulting data, which is summarized in analysis notebooks available on this GitHub repository.
How are different willow species related?
Understanding the historical evolutionary relationships between willow species has challenged researchers for hundreds of years, because willows have had periods of rapid diversification, and because species hybridize readily. Past attempts at using genetic markers to understand species relationships have ended in “spectacular failures”. Another major research goal for our group is to use the new targeted sequence capture array that I have designed to attempt to better understand the relationships among willow species. This is important to us, because knowing how species are related to each other, how they have diverged historically, is a critical context for understanding the evolution of their sex determining genes.
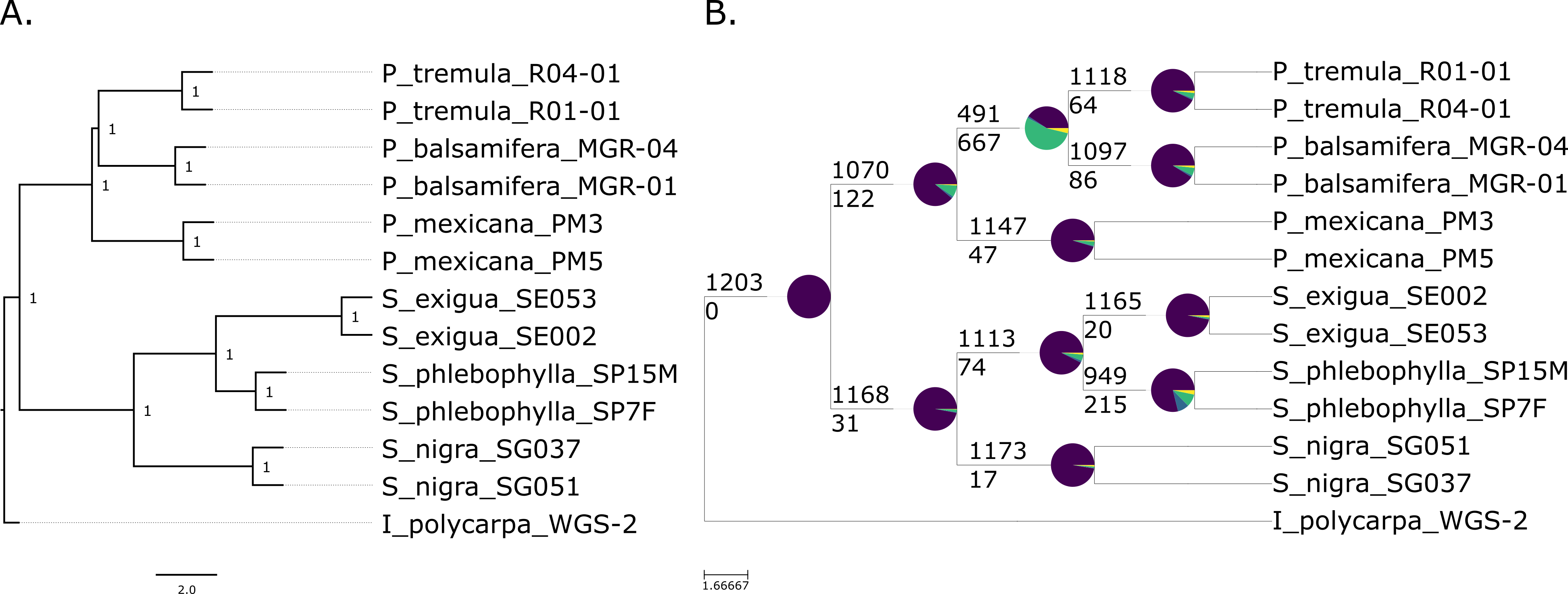 A sample
phylogeny of willows and poplars prepared with targeted sequence capture.
Both trees were prepared with data from 1,203 genes. The tree on the left
shows the relationships among the species. The pie charts in the tree on the
right shows the number of genes that support that relationship (purple) versus
alternative relationships that are not displayed (other colors).
A sample
phylogeny of willows and poplars prepared with targeted sequence capture.
Both trees were prepared with data from 1,203 genes. The tree on the left
shows the relationships among the species. The pie charts in the tree on the
right shows the number of genes that support that relationship (purple) versus
alternative relationships that are not displayed (other colors).
In a recent paper, available as a pre-print on bioRχiv I report a trial phylogeny that I prepared using the targeted sequence capture array that I developed. This was a small proof of concept that I prepared to report on the methods that we are implementing for a larger phylogenetic analysis currently underway, with sequence data from ~180 individuals representing the major groups of species in both genera. We are hopeful that this resource will provide greater insight into the complex evolutionary history of these species. I published analysis notebooks that detail the pipeline I developed for this analysis on this GitHub repository.
Using patterns of gene expression to understand complex traits
In addition to the experiments outlined above using data related to the location of genes within the genome (sequences of DNA), we also use patterns of gene expression (sequences of RNA) to determine how organisms develop different sexual traits, and how they cope with stressful environments. Sequencing RNA (“RNA-seq”) from individuals that differ in some trait of interest, or before and after inducing a stressful treatment, allows us to see how the overall expression of genes changes.
How do plants make different types of flowers?
The vast majority of plants are hermaphrodites (all individuals make both pollen and seeds), yet a small number of species do have separate male (pollen only) and female individuals (seeds only), notably all willow and poplar species. While we are still working to understand what specific genes guide the development of sexual traits, even without knowing this we can compare the overall differences in the expression of all genes between males and females to understand which genes are likely to be important in the proper development of the differences in traits between the sexes.
 Differential gene
expression in male and female flowers of balsam poplar. Each row is a single
gene and each column is an individual male (M1-M5) or female (F1-F5) tree.
The color represents the variation in gene expression values: cooler colors are
genes with low expression and warmer colors are genes with expression (i.e.
number of copies of RNA from that gene that were present in the sample).
Differential gene
expression in male and female flowers of balsam poplar. Each row is a single
gene and each column is an individual male (M1-M5) or female (F1-F5) tree.
The color represents the variation in gene expression values: cooler colors are
genes with low expression and warmer colors are genes with expression (i.e.
number of copies of RNA from that gene that were present in the sample).
In this paper published in the journal New Phytologist I used an analysis of RNA-seq data to show that males and females of the tree species balsam poplar differ mostly in the expression of genes in their flowers, but not their leaves. The genes that I identified here include some promising candidates for both the master sex-determination genes in this species, as well as downstream targets of those genes.
How do plants respond to freezing temperatures?
Cold weather can be very damaging to plants, both because the water within the plant tissue can freeze causing damage to tissues, and also because water in the environment freezes and becomes unavailable like in a drought.
 Differential gene
expression before and after exposure to cold temperatures in
Arabidopsis thaliana mutant lines. Each row is a single
gene (names at right) and each column is a plant line.
The color represents the variation in gene expression values before and after
cold treatment. Cooler colors show higher expression before cold treatment,
warmer colors show higher expression after cold treatment (I agree that this
is confusing, I lost this argument with a colleague on this study).
Differential gene
expression before and after exposure to cold temperatures in
Arabidopsis thaliana mutant lines. Each row is a single
gene (names at right) and each column is a plant line.
The color represents the variation in gene expression values before and after
cold treatment. Cooler colors show higher expression before cold treatment,
warmer colors show higher expression after cold treatment (I agree that this
is confusing, I lost this argument with a colleague on this study).
In this paper published in the journal American Journal of Botany I used the model plant species mouse-eared cress (Arabidopsis thaliana) to determine what changes occurred in gene expression before and after freezing temperatures. The plants in this study came from two natural populations in Italy and Sweden that vary how the length and severity of the freezing period of winter. Past studies have shown that plants in both populations grow and survive better than they do if they are transplanted into each other’s environments, and this difference has been found to be caused by a single gene that does not function properly in the Italian plants. With plants from these natural populations, we used a combination of traditional plant crossing, as well as CRISPR/Cas9, to create mutant lines that varied for only the single gene that we expected to be the major factor that causes differences in survival through freezing. After subjecting the mutant lines to cold temperatures, we found that there were 10 genes that showed significant differences before and after cold treatment. These genes are promising candidates for understanding the mechanism that plants use to survive through cold winter temperatures.
Reproductive behaviors in plants
 Mapping wild plant populations near Simmonsville, VA.
Mapping wild plant populations near Simmonsville, VA.
Sexual reproduction typically requires the interaction of two individuals, and so the demographics of a population can contribute to whether an individual can successfully reproduce. In my dissertation research I focused on the plant species Silene vulgaris, because in this species some individuals are capable of “selfing,” or reproducing without a partner (hermaphrodites), while others depend on interactions with others in order to reproduce (females). I used both wild populations as well as controlled gardens I constructed to determine how different frequencies of both types of plants could alter the reproductive success of individuals.
How are individuals interacting within populations?
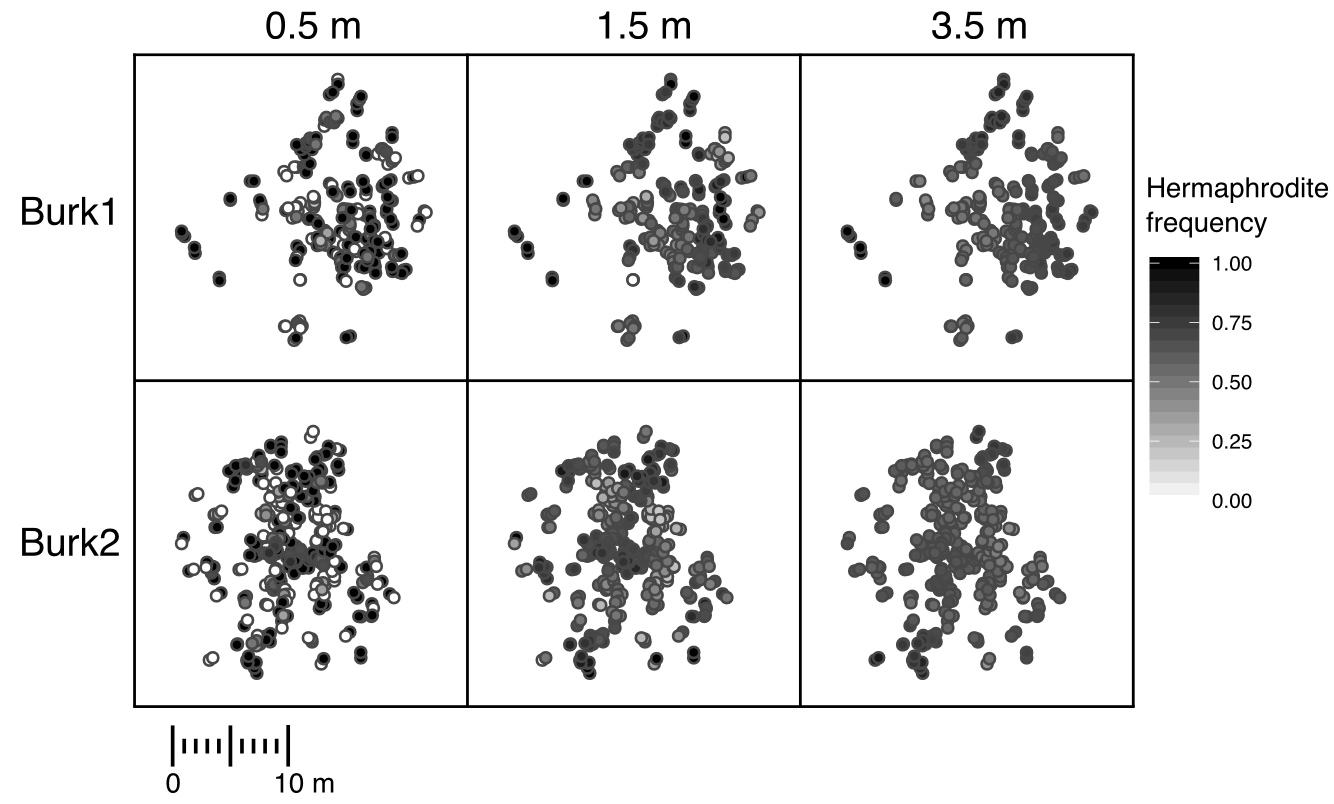 The demography of two populations of Silene vulgaris.
Each point represents and individual, and the shading of the point represents
the subjective hermaphrodite frequency at that scale.
The demography of two populations of Silene vulgaris.
Each point represents and individual, and the shading of the point represents
the subjective hermaphrodite frequency at that scale.
In this study published in the journal Ecology and Evolution I found that there is continuous variation at a fine scale within populations of S. vulgaris. Understanding whether this variation matters depends on knowing how far pollen is carried by pollinators. If it is only carried short distances (~0.5 meters), then there is a great amount of variation in pollen availability within the population, while if pollinators travel farther (1.5-3.5 meters), most individuals experience the same availability of pollen. This result led me wonder…
Does the behavior of pollinators differ depending on demographics of the population?
 Eagerly anticipating pollinators
Eagerly anticipating pollinators
Because plants can’t move, their social environment in terms of reproduction is
determined by the behavior of pollinating insects. If pollinators
exhibit strong preferences for the sex of plants, or the sex ratio of
aggregations of plants, then those preferences can dramatically impact the
reproductive success of individual plants. In this study I used
experimental plots of Silene vulgaris to test the prediction that the
frequency of hermaphrodites in the plot would affect the recruitment and
behavior of pollinators. S. vulgaris is pollinated both by bees and flies
during the day, and also by moths during the night.
 The probability an insect visited an individual (A & B), the number of insects that visited an individual (C & D), and the duration of visits to that individual (E & F) during morning and evening observations.
The probability an insect visited an individual (A & B), the number of insects that visited an individual (C & D), and the duration of visits to that individual (E & F) during morning and evening observations.
We found that, although there were differences between daytime and nighttime pollinators, neither group of pollinators behaved differently among the sex ratio treatments. This suggests that the fine-scale variation in sex ratio in plant populations are likely to have strong effects on the reproductive success of individuals (more like the 0.5m scale).